


Todos nós vimos aquele momento em um programa de TV policial em que um detetive está analisando imagens de segurança granuladas e de baixa resolução, localiza uma pessoa de seu interesse na fita e pede indiferentemente a um técnico de CSI para “melhorar isso”. Alguns cliques no teclado depois e voilà – eles têm uma imagem perfeita e clara do rosto do suspeito. Isso, é claro, não funciona no mundo real, como muitos críticos de cinema e pessoas na internet gostam de apontar.
No entanto, cientistas da vida real desenvolveram recentemente uma verdadeira ferramenta de “aprimoramento”: uma que melhora a resolução e a precisão de microscópios poderosos que são usados para revelar percepções em biologia e medicina.
Em um estudo publicado na Nature Methods, uma equipe multiinstitucional liderada por Tom Terwilliger do New Mexico Consortium e incluindo pesquisadores do Lawrence Berkeley National Laboratory (Berkeley Lab) demonstra como um novo algoritmo de computador melhora a qualidade da estrutura molecular 3-D mapas gerados com microscopia crioeletrônica (crio-EM).
Por décadas, esses mapas crio-EM – gerados por muitas imagens microscópicas e aplicação de software de processamento de imagens – têm sido uma ferramenta crucial para pesquisadores que buscam aprender como funcionam as moléculas dentro de animais, plantas, micróbios e vírus. E, nos últimos anos, a tecnologia crio-EM avançou a ponto de poder produzir estruturas com resolução em nível atômico para muitos tipos de moléculas. No entanto, em algumas situações, mesmo os métodos crio-EM mais sofisticados ainda geram mapas com resolução mais baixa e maior incerteza do que o necessário para desvendar os detalhes de reações químicas complexas.
“Em biologia, ganhamos muito conhecendo a estrutura de uma molécula”, disse o co-autor do estudo Paul Adams, diretor da Divisão de Biofísica Molecular e Bioimagem Integrada do Laboratório de Berkeley. “As melhorias que vemos com este algoritmo tornarão mais fácil para os pesquisadores determinarem modelos estruturais atomísticos de dados de crio-microscopia eletrônica. Isso é particularmente importante para a modelagem de moléculas biológicas muito importantes, como aquelas envolvidas na transcrição e tradução do código genético, que são frequentemente vistos apenas em mapas de resolução mais baixa devido às suas estruturas multi-unidades grandes e complexas.”
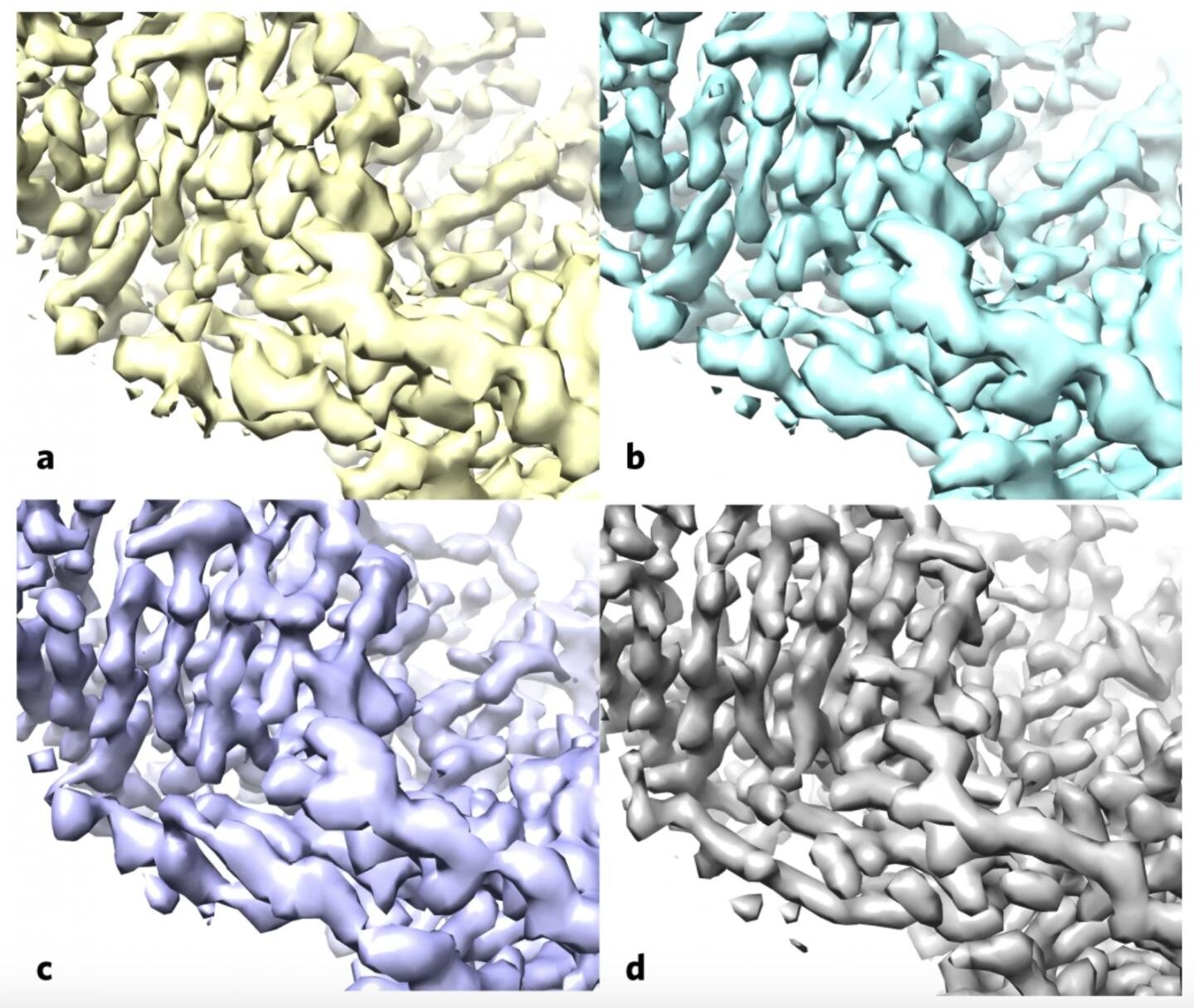
O algoritmo aprimora os mapas moleculares filtrando os dados com base no conhecimento existente de como as moléculas se parecem e como melhor estimar e remover o ruído (dados indesejados e irrelevantes) nos dados de microscopia. Uma abordagem com a mesma base teórica foi usada anteriormente para melhorar os mapas estruturais gerados a partir da cristalografia de raios-X, e os cientistas já propuseram seu uso em crio-EM. Mas, de acordo com Adams, ninguém foi capaz de mostrar evidências definitivas de que funcionava para o crio-EM até agora.
A equipe – composta por cientistas do New Mexico Consortium, Los Alamos National Laboratory, Baylor College of Medicine, Cambridge University e Berkeley Lab – aplicou primeiro o algoritmo a um mapa publicamente disponível da proteína humana apoferritina que é conhecida por ter 3,1 angstrom resolução (um angstrom é igual a 10 bilionésimos de um metro; para referência, o diâmetro de um átomo de carbono é estimado em 2 angstroms). Em seguida, eles compararam sua versão aprimorada com outro mapa de referência de apoferritina disponível publicamente com resolução de 1,8 angstrom e encontraram correlação aprimorada entre os dois.
Em seguida, a equipe usou sua abordagem em 104 conjuntos de dados de mapas do Banco de Dados de Microscopia Eletrônica. Para uma grande proporção desses conjuntos de mapas, o algoritmo melhorou a correlação entre o mapa experimental e a estrutura atômica conhecida e aumentou a visibilidade dos detalhes.
Os autores observam que os benefícios claros do algoritmo em revelar detalhes importantes nos dados, combinados com sua facilidade de uso – é uma análise automatizada que pode ser realizada em um processador de laptop – provavelmente fará parte de uma parte padrão do fluxo de trabalho cryo-EM avançando. Na verdade, Adams já adicionou o código-fonte do algoritmo ao conjunto de software Phenix, um pacote popular para solução de estrutura macromolecular automatizada para o qual ele lidera a equipe de desenvolvimento.
Esta pesquisa foi parte dos esforços contínuos do Berkeley Lab para avançar os recursos da tecnologia crio-EM e para ser o pioneiro em seu uso para descobertas científicas básicas. Muitas das invenções revolucionárias que permitiram o desenvolvimento do crio-EM e mais tarde levaram-no à sua excepcional resolução atual envolveram cientistas do Berkeley Lab.
Achou útil essa informação? Compartilhe com seus amigos! 
Deixe-nos a sua opinião aqui nos comentário.